BLAST是“局部相似性基本查询工具”(Basic Local Alignment Search Tool )的缩写。 这是美国生物技术信息中心(NCBI )开发的基于序列相似性的数据库检索程序。 该程序将DNA/蛋白质序列与公开数据库的所有序列进行一致比对,找出类似序列。
生物序列的相似性
相似性(similarity ) :是指非常直接的数量关系,如部分相同或相似的百分比或其他适当的测量值。 例如,a序列和b序列的相似性为80%,或4/5。 这是定量关系。 当然,可以进行自身的局部比较。
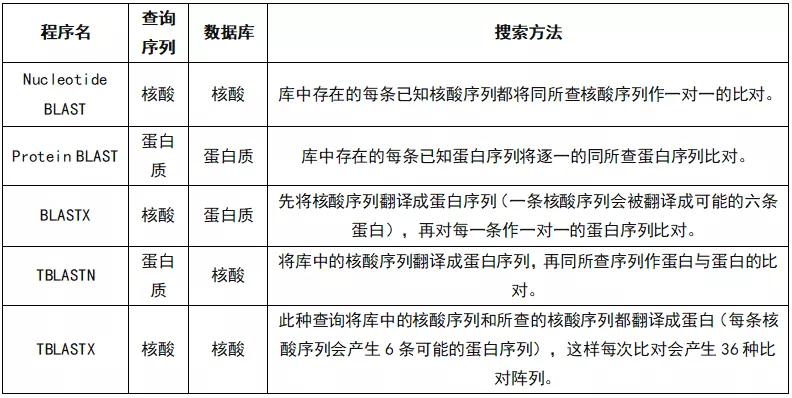
序列相似性比较:用于将研究对象的序列与DNA或蛋白质序列库进行比较,决定该序列的生物学特性。 也就是说,我得出与这个序列类似的已知序列是什么。 为了完成这项工作,需要使用两个序列比较算法。 常用的程序有BLAST、FASTA等。
Blast的主要程序
1 .打开blast (https://blast.NCBI.NLM.NIH.gov/blast.CGI )。
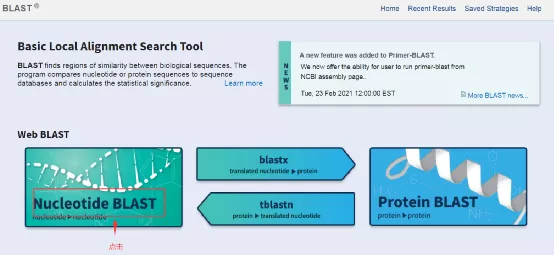
2 .输入归类序列和参数
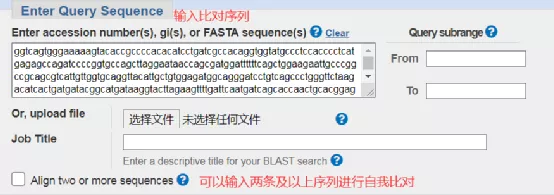
3 .分析3. BLAST结果
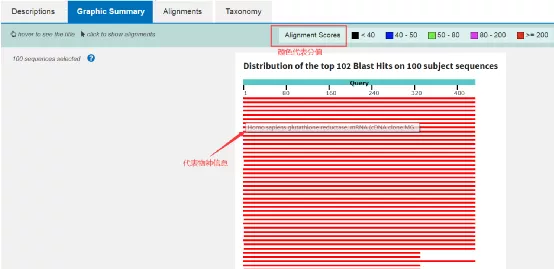
一般来说,评价一个blast结果的标准主要有三个项目,e值(Expect )、一致性)、缺失或插入) Gaps )。 一般情况下,还会添加序列长度(length )。
Score (序列比对中计算的得分值,得分越高序列比对结果越好。
Expect :表示随机匹配的可能性。 e的值越小,序列越相似,e的值越大,随机匹配的可能性也越大。 e的值为零或接近零时,基本上完全一致。
(Identities )序列相似性、匹配上碱基数占总序列长度的百分率。
GPS :插入或缺失。 用“—”表示。
是的,这次推送的有关NCBI的三篇文章都解释完了
每个都是:
目的基因检索
QCR引物设计
基因序列比对
记住了吗?